Development of PCR & Sanger Sequencing
Polymerase Chain Reaction
As you have already learned in Introductory Biology, Polymerase Chain Reaction (PCR), is a powerful molecular biology technique used to amplify specific DNA sequences. Developed in the 1980s by Kary Mullis, PCR revolutionized molecular biology by enabling rapid and efficient amplification of DNA, even from minute quantities of starting material. Before the development of PCR, DNA amplification was a laborious and time-consuming process that often involved cloning DNA fragments into bacterial vectors and culturing them in bacterial cells. Mullis’ invention of PCR provided a simple and efficient alternative, allowing researchers to amplify DNA in a test tube through a series of repeated temperature cycles.
Principle of PCR
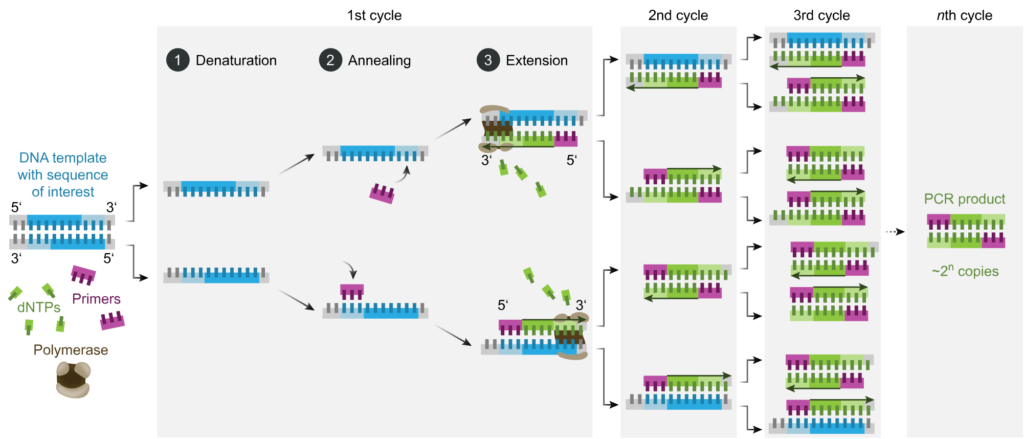
PCR amplifies specific DNA sequences through a series of three basic steps:
- Denaturation: The DNA template is heated to a high temperature (typically 94-98°C), causing the double-stranded DNA to denature into single strands.
- Annealing: The reaction temperature is lowered to allow primers (short DNA sequences complementary to the target DNA) to bind to their complementary sequences on the template DNA.
- Extension: The reaction temperature is raised, and a DNA polymerase enzyme synthesizes new DNA strands by extending from the primers along the template DNA. This process generates complementary copies of the target DNA sequence.
By repeating these temperature cycles multiple times (usually 20-40 cycles), PCR exponentially amplifies the target DNA sequence, resulting in millions to billions of copies of the desired DNA fragment.
Applications of PCR:
PCR has numerous applications in molecular biology, genetics, forensics, and medical diagnostics, including:
- Gene cloning and DNA sequencing
- Gene expression analysis (RT-PCR)
- DNA fingerprinting and forensic analysis
- Pathogen detection and microbial identification
- Environmental DNA (eDNA) analysis
Di-deoxyribonucleotide (‘dideoxy’) chain-termination sequencing
In the late 1970s, Frederick Sanger and his colleagues at the Medical Research Council Laboratory of Molecular Biology in Cambridge, UK, developed a groundbreaking method for sequencing DNA known as dideoxy sequencing, or Sanger sequencing. This method represented a significant advancement in molecular biology, offering a reliable and efficient means of determining the sequence of nucleotide bases in a DNA molecule. Before the advent of dideoxy sequencing, DNA sequencing was a cumbersome and time-consuming process. Early methods, such as Maxam-Gilbert sequencing, relied on chemical treatments to break DNA at specific nucleotide positions and infer the sequence indirectly. These methods were labor-intensive and often yielded limited amounts of sequence data. Dideoxy sequencing revolutionized DNA sequencing by introducing a new approach based on the incorporation of chain-terminating dideoxynucleotides (ddNTPs) during DNA synthesis. Unlike normal deoxynucleotides (dNTPs), which contain a 3′ hydroxyl group required for chain elongation, ddNTPs lack this group, leading to termination of DNA synthesis when they are incorporated into the growing DNA strand. In dideoxy sequencing, the DNA template is first denatured and annealed with a short DNA primer. DNA synthesis is then initiated using DNA polymerase and a mixture of normal dNTPs and a small amount of ddNTPs labeled with fluorescent dyes. As DNA synthesis proceeds, ddNTPs are randomly incorporated into the growing DNA strand, leading to chain termination at specific nucleotide positions.
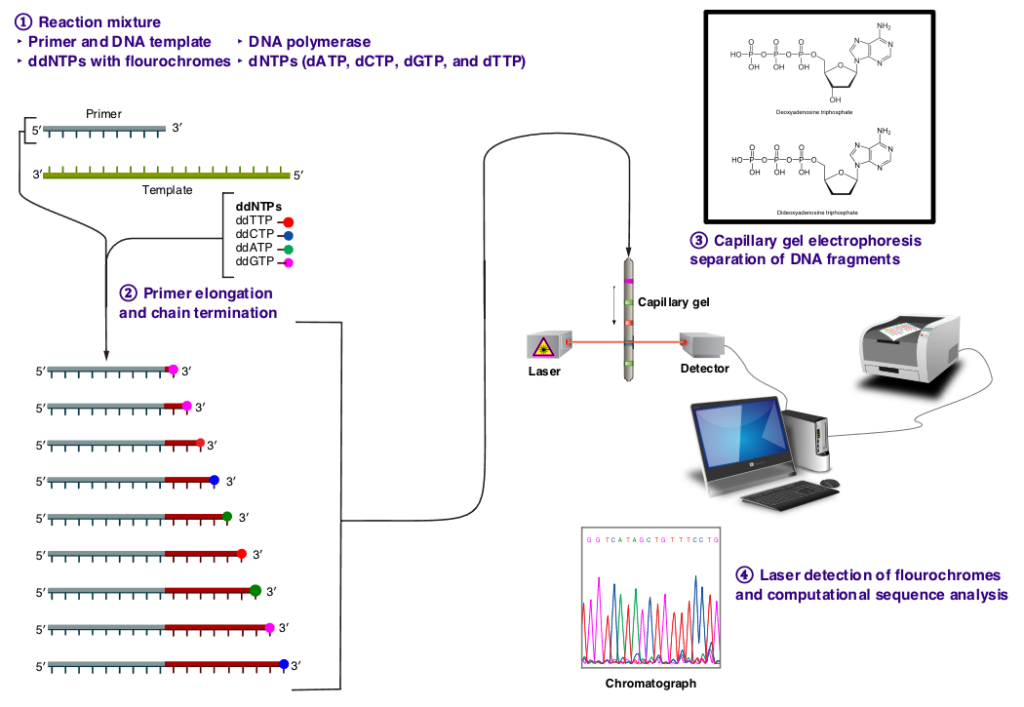
Dideoxy sequencing had a profound impact on molecular biology and genetics, enabling high-throughput DNA sequencing with unprecedented accuracy and reliability. Its development paved the way for large-scale genome sequencing projects, such as the Human Genome Project, which aimed to sequence the entire human genome. Moreover, dideoxy sequencing facilitated the development of various molecular markers, including microsatellites, single nucleotide polymorphisms (SNPs), and restriction fragment length polymorphisms (RFLPs). These markers have become essential tools in molecular ecology and evolutionary biology, allowing researchers to study genetic diversity, population structure, and evolutionary relationships at the molecular level.
PCR & Sanger Sequencing: The Dynamic Duo
The combination of PCR with Sanger sequencing revolutionized molecular ecology and evolutionary biology by enabling rapid and accurate DNA sequencing of specific target regions. Researchers could now amplify target DNA sequences of interest using PCR and then sequence them using Sanger sequencing, providing detailed information about genetic variation, population structure, and evolutionary relationships. This approach facilitated the development of various molecular markers, such as microsatellites, SNPs, and mitochondrial DNA sequences, which have become indispensable tools in molecular ecology and evolutionary biology. These markers allow researchers to study genetic diversity, population dynamics, gene flow, and evolutionary processes in natural populations with unprecedented resolution and precision. Overall, the integration of PCR and Sanger sequencing has had a transformative impact on molecular ecology and evolutionary biology, enabling groundbreaking research and advancing our understanding of the genetic basis of biodiversity, adaptation, and evolution in natural systems.
PCR-based markers rely on the Polymerase Chain Reaction (PCR) to amplify specific DNA sequences of interest. This technique allows for rapid and efficient amplification of target DNA sequences, enabling high-throughput analysis of genetic variation. PCR-based markers are highly sensitive and specific, with the ability to amplify DNA from minute quantities of starting material and detect single nucleotide differences. Examples of PCR-based markers include microsatellites, Single Nucleotide Polymorphisms (SNPs), Amplified Fragment Length Polymorphisms (AFLPs), Restriction Fragment Length Polymorphisms (RFLPs), and mitochondrial DNA (mtDNA) sequences. PCR-based markers are widely used in genetics, genomics, forensics, medical diagnostics, and molecular ecology due to their versatility and scalability.
Non-PCR-based markers on the other hand, do not involve PCR amplification and rely on alternative methods for detecting genetic variation. These markers may include variations in DNA sequence, protein products, or chromosomal structure. While non-PCR-based markers may offer certain advantages such as simplicity and lower cost, they may also have limitations in terms of sensitivity, specificity, and resolution. Examples of non-PCR-based markers include allozymes, restriction site polymorphisms, karyotypes, and isozymes. Non-PCR-based markers have been widely used in molecular ecology, population genetics, and evolutionary biology, providing valuable insights into genetic diversity and evolutionary processes.
Table 2. Summary of differences between PCR and Non-PCR based molecular markers
| Aspect | PCR-Based Markers | Non-PCR-Based Markers |
| Methodology | Amplify specific DNA sequences using PCR | Do not involve PCR amplfication |
| Amplification | Exponential amplification of target DNA sequences | No amplification step involved |
| Speed | Rapid amplification within hours | Time-consuming, may take days or weeks |
| Sensitivity | Highly sensitive, can amplify DNA from minute quantities of starting material | Less sensitive, may require larger amounts of starting material |
| Specificity |
Highly specific, amplifies only target DNA sequences with complementary primers | Less specific, may capture non-target DNA sequences |
| Accuracy | High accuracy, low error rate | Accuracy may vary depending on the method |
| Throughput | High throughput, allows for simultaneous amplification of multiple targets | Lower throughput, may analyze fewer loci simultaneously |
| Applications | Widely used in genetics, genomics, forensics, medical diagnostics, and molecular ecology | Commonly used in molecular ecology, population genetics, and evolutionary biology |
| Examples | Microsatellites, SNPs, AFLPs, RFLPs, mtDNA sequences | Allozymes, restriction site polymorphisms, karyotypes, isozymes |
| Cost | Moderate to high, depending on the number of markers and samples | Lower cost for some methods, but may require specialized reagents or equipment |
| Data Analysis | Requires sequencing or fragment analysis after PCR amplification | Direct analysis of electrophoretic patterns or sequencing data |
| Resolution | High resolution, can detect single nucleotide differences and subtle genetic variations | Resolution may vary depending on the method, may not detect single nucleotide differences |
| Flexibility | Highly flexible, allows customization of primer sequences and amplification conditions | Less flexible, dependent on available markers and methods |
| Ease of Use | Requires specialized equipment and expertise in PCR techniques | May be simpler and require less specialized equipment |
Media Attributions
- PCR © Enzoklop is licensed under a CC BY-SA (Attribution ShareAlike) license
- sangersequencing © Estevezj is licensed under a CC BY-SA (Attribution ShareAlike) license

{kind=link}
{kind=link}